Download

1 / 25
270 likes | 845 Views
Usher Syndrom. Referentin: Mareen Robitzsch. *Das Usher Syndrom (USH)*. nach dem englischen Augenarzt Charles H. Usher (1914) Schwerhörigkeit/ Gehörlosigkeit und die Degeneration der Netzhaut im Auge (Retinitis Pigmentosa, RP). *Das menschliche Ohr*. * Aufbau des Ohrs *.

E N D
Usher Syndrom Referentin: Mareen Robitzsch
*Das Usher Syndrom (USH)* • nach dem englischen Augenarzt Charles H. Usher (1914) • Schwerhörigkeit/ Gehörlosigkeit und die Degeneration der Netzhaut im Auge (Retinitis Pigmentosa, RP)
*Aufbau des Ohrs* • Man teilt das Ohr in drei Bereiche ein
*Die akustische Wahrnehmung* • Als auditive oder akustische Wahrnehmung (auch Gehörsinn/Hörsinn) bezeichnet man die Stimulation eines Sinnesorgans durch Luftdruckveränderungen.
*Haarzellen* • Auf der Basilarmembran sind vier Reihen von Haarzellen angeordnet: • äußeren Haarzellen (3 Reihen) Verstärkung der Schallwanderwellen innerhalb der Cochlea (Cochlear-Amplifier, cochleärer Verstärker) • inneren Haarzellen (1 Reihe) Umwandlung mechanischer Schwingungen in Nervenimpulse ("Transduktion„)
*Tonhöhenwahrnehmung* • durch die Wanderwelle der Cochlea und dessen maximaler Amplitude • der Ort der maximalen Amplitude ist abhängig von der Frequenz der Wanderwelle (der Tonhöhe) • im Gehirn wird die Stärke und der Ort der Haarzellenreizung ausgewertet • das Gehirn konstruiert Lautstärke und Tonhöhe des ursprünglichen Schallreizes
* “Tip-Links“ * • dünne Proteinfäden, • verbinden jede Stereozilie mit ihrer nächst längeren und nächst kürzeren Nachbarin • es entstehen Ketten von Stereozilien, von der Kürzesten zur Längsten verlaufend • man vermutet, dass Tip-Links die Öffnung der Transduktionskanäle kontrollieren • bei der Entfernung von Tip-Links verlieren die Haarzellen ihre Mechanosensitivität
*Die Funktion der „Tip-Links“* • Bei Auslenkung des Haarbündels verschieben sich die benachbarten Stereozilien gegeneinander. • Die Tip-links werden gestrafft und üben eine Zugkraft auf die Plasmamembranen der beiden Stereozilien aus. • Diese Kraft öffnet die Transduktionskanäle und löst damit die Depolarisation der Zelle aus.
*Stäbchen und Zapfen* • Zwei Typen von Fotorezeptoren: • Stäbchen- besonders lichtempfindlich- auch in der Dämmerung aktiv (Dunkeladaption) • Zapfen- dienen zur Farbwahrnehmung und zum Tagessehen
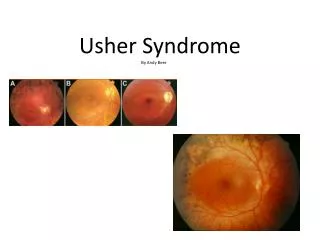
*Retinitis Pigmentosa (RP)* • Das Usher Syndrom (USH) ist eine Form der Retinitis Pigmentosa (RP), einer vererbbaren, degenerativen Augenerkrankung, die in der Kindheit oder im frühen Erwachsenenalter auftritt und bisher nicht heilbar ist. • Bei RP sterben die Netzhautzellen allmählich oder schubweise ab. • Es tritt eine Einschränkung des Gesichtsfeldes auf, der „Tunnelblick“ und/oder Gesichtsfeldausfälle (Skotomen). • Weitere Merkmale der RP sind: • Nachtblindheit (fehlende Hell-Dunkel-Adaption) • Blendungsempfindlichkeit • Störung des Farb- und Kontrastsehens
*Krankheitsverlauf von RP* • Vom Rand der Netzhaut zum Zentrum fortschreitend kommt es zur Einlagerung von Pigmentkörperchen (Lichtundurchlässigkeit). • Nachtblindheit (Absterben der Stäbchen) • Vom peripheren Bereich ausgehend beginnt die Gesichtsfeldeinschränkung- „Röhrengesichtsfeld“ oder „Tunnelblick“. (8.-17.Lebensjahr, weiter 20.-30. Lebensjahr- Zerfall der Zapfen) • Erblindung in Verbindung mit Katarakt (grauer Star), Glaukom (grüner Star) oder Myopie (Kurzsichtigkeit), (ab dem 40. Lebensjahr)
*Krankheitsbild* • USH Typ1: Gehörlosigkeit und Gleichgewichtsstörungen von Geburt an, später kommt RP hinzu (Häufigkeit 33-44%) • Untergruppen A-G, am häufigsten USH 1B • USH Typ2: hochgradige Schwerhörigkeit, tritt nicht zwangsläufig mit Gleichgewichtsstörung und RP auf (Häufigkeit 56-67%) • Subtypen A und B, am häufigsten USH 2A • USH Typ3: von Geburt an mittelgradige, sich verstärkende Schwerhörigkeit und RP (sehr seltene Form, nur in Finnland aufgetreten)
*Ursachen für beobachtete Verlaufsvariabiliät* • unterschiedliche primäre Gendefekte • verschiedene Allele des selben Gens • unterschiedliche Umweltbelastungen
*Mutation des MYO7A-Gens* • Bei USH 1B resultiert aus der Mutation des MYO7A- Gens die häufigste Form der kombinierten Blindheit und Gehörlosigkeit. • MYO7A verschlüsselt das Protein Myosin 7a. • Die Rolle des Proteins ist unbekannt, es wird vermutet, dass es eine Transportfunktion innerhalb der Photorezeptorzellen (RP) und der Zellen des retinalen Pigmentepithels (RPE) ausübt.
*Erbschema zur Erklärung des Usher Syndrom**autosomal rezessive Vererbung*
*Therapieansätze* • Morphologie: • Netzhauttransplantation • Molekulargenetik: • Lokalisierung von Genen • Als erfolgsversprechend gelten: • pharmakologische Behandlung • Austausch des mutierten Gens durch Einschleusen von Vektor-Viren • Netzhauttransplantation • Einsatz von Prothesen (Neuroprothetik) • Cochlear Implant (CI)
*Behandlung des USH* • Einnahme von Vitamin A • optische Sehhilfen • Operation des Cataract • In Deutschland leben schätzungsweise 5.000 Betroffene
*Quellen* • Usher-Homepag: www.dbcent.dk/text_version/Ushertxt.htm www.ushernet.org www.blindness.org/usher/ • Literatur: • Rosemarie Große-Wilde: • User-Syndrom. Was ist das? Pro-Retina-Info-Serie Nr. 4, 1-2005 (4. Auflage) • Kirsten Rappmund: Jubiläumsschrift zum 15jährigen Bestehen des AK Usher (Zusammenstellung von Aufsätzen, Seminarprogrammen und –Berichten 199-2006) • Rosemarie Große-Wilde: • Anregungen und Tips für USHER – Betroffenen • Hilfen bei fortschreitender Hör- und Sehschädigung. • Pro-Retina-Sonderdruck Nr. 108, 3/1998. • Roger Reichardt: • Das User-Syndrom, Diplomarbeit, Berlin 1999