Download

1 / 140
1.42k likes | 1.67k Views
MBV3070 Bioinformatikk; proteiner. Lærer: Vincent Eijsink, Institutt for Kjemi, Bioteknologi og Matvitenskap, Norges landbrukshøgskole; Telefon: 64949472; E-post: Vincent.eijsink@ikbm.nlh.no; Web: kitin.nlh.no/enzymgruppa Pensum:

E N D
MBV3070 Bioinformatikk; proteiner Lærer: Vincent Eijsink, Institutt for Kjemi, Bioteknologi og Matvitenskap, Norges landbrukshøgskole; Telefon: 64949472; E-post: Vincent.eijsink@ikbm.nlh.no; Web: kitin.nlh.no/enzymgruppa • Pensum: Tatt fra Lesk, side 31-53, 115-156, 187-196 og 216-271, med fokus På kapittel 5 (s 216-271) og utvidet med tilleggsinformasjon fra andre kilder. Pensumet består av: • Det som behandles under forelesningene (Lysbildene) • Øvelsene • Guex et al. Protein modelling for all TIBS 24:364-367 (1999) (om SwissModel) • Schonbrun et al., Protein structure prediction in 2002, Current Opinion in Structural Biology, 12:348-354 (2002) Les boka! Det er bra skrevet.
MBV3070 Bioinformatikk; proteiner Program: • Ti 20.04: forelesninger • Fre 30.04: forelesninger • Ti 04.05: forelesninger; øvelsene deles ut • Fre 07.05: forelesninger; diskusjon av de artikler som er pensum • Ti 18.05: gjennomgang av øvelsene; spørsmål, oppsummering
Tema • Protein struktur og folding, oversikt (repetisjon) • Strukturbestemmelse av proteiner • Protein struktur databaser (PDB og andre) • Visualisering av strukturer • Sammenligning og kategorisering av av strukturer • Multiple sequence alignments (MSAs) • Databaser av MSAs • Sekvens-basert prediksjon av funksjon, evolusjonær opphav, struktur, fysiske egenskaper • Fold recognition (”threading”) • Prediksjon av sekundær og tertiær struktur; bygging av tre-dimensjonale protein modeller • Proteomics, structural genomics • Bruk av proteinkunnskap i annotering av genomer • Avansert protein bioinformatikk (f eks molekylær dynamika, elektrostatika, protein design – stort sett bare eksempler)
Generelt om proteiner • Matthews, Van Holde & Ahern (KJB201) eller andre generelle biokjemi tekstbøker • C. Branden & J. Tooze, Introduction to Protein Structure (ISBN 0-8153-2305-0) • A. Lesk, Introduction to Protein Science (ISBN 0-19-926511-9) • G.A. Petsko, D. Ringe, Protein Structure and Function (ISBN: 0878936637)
Andre nyttige bioinformatikk bøker • Mount, D., Bioinformatics – Sequence and genome analysis (www.bioinformaticsonline.org) • Baxevanis, A.D. & Ouellette, B.F., Bioinformatics. A practical guide to the analysis of genes and proteins De som vil vite mer om model-bygging kan lese: Rodriguez & Vriend, ”Professional gambling”, http://www.cmbi.kun.nl/gv/articles/text/gambling.html (en detaljert og relativ enkel forklaring av prinsippene av ”model-building- by- homology” som programmet WHAT IF gjør det)
Bioinformatikk = modellering av protein struktur? Se C. Levinthal, Scientific American, juni 1966: ”Molecular Model-building by computer” (ikke pensum)
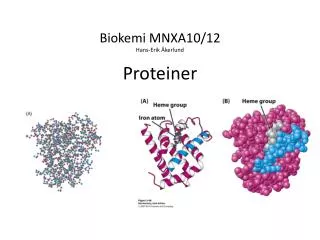
Protein struktur; terminologi • Primærstruktur: Aminosyrerekkefølge • Sekundærstruktur: Faste strukturelementer, karakterisert av standard type hydrogen bindinger mellom hovedkjede atomer: a-helix, b-sheet, turn, coil mfl. (ofte deler man i bare tre varianter: a-helix, b-sheet, rest) • Tertiær struktur: Hele strukturen, inklusive alle detaljer (= ”tertiære interaksjoner”) • Kvarternær struktur: multimere proteiner; hver subunit har sin tertiær struktur; hele komplekset har en kvarternær struktur
Amino acids OBS! Bilder av proteinstrukturer viser som oftest ikke hydrogen atomer
Amino acids Det finnes mange måter for å gruppere aminosyrer på, og dette er ikke triviell (jfr f eks sekvenssammenligning)
Alanine Oksygen Nitrogen Karbon Hydrogen Glycine (R = H) og proline (R kovalent bundet til N) er spesial
Stereokjemi: CORN-regelen All amino acids found in proteins encoded by the genome have the L-configuration at this chiral centre. This configuration can be remembered as the CORN law. When read clockwise, the groups attached to the Calpha spell the word CORN.
Peptidbindingen er plan R O O- - N – C - -+N = C - H H H C-a C-a N H R H
Rotasjonsakser i peptider R R H H H y f N C-a C-a C-a N H R H Phi og Psi begrenses av generell kjemiske prinsipper (”staggered” er bedre enn ”eclipsed”); variasjon i R påvirker mulighetene.
Torsion Angles • Ingen vesentlig rotasjon om omega. • Bindingsvinkler og lengder viser ingen vesentlig variasjon • Phi & Psi avgjør strukturen
Ramachadran plot • Ramachadranplott viser kombinasjoner av f og y som fore-kommer i protein strukturer • Dette er dermed ener-getisk gunstige kombi-nasjoner av f og y • I slike plots skiller glysin og proline seg ut
Sekundær struktur (side 40, Fig. 1.7) • Strukturelementer med karakteristiske mønstre med • hydrogenbruer: • alpha-helix • 310 helix • b-flak (sheet, strand) • Diverse typer ”turns” Ofte snakker man bare om 3 ”states”: helix, strand, coil
Properties of the alpha-helix • Regelmessig hydrogenbru mønster • ”Overskudd” av N-H på N-terminus og av C=O på C-terminus -> Dipol
En a-helix er ofte amfipatisk: Helical wheel
b-strand Sidegruppene peker vekselvis ut på hver side av beta-flaket; en enkel beta-strand kan også være amfipatisk En beta-”strand” har karakteristiske phi og psi vinkler. Flere beta-strands kan gå sammen og danne en beta-”sheet”
Vesentlig forskjell mellom a og b: • Helikser stabiliseres av interaksjoner som er ”lokal” i sekvensen. • b-sheets stabiliseres av interaksjoner mellom hovedkjedeatomer som kan ligge langt fra hverandre i sekvensen (Hvilke av disse to typer struktur vil være letter å predikere på basis av kun aminosyresekvensen?)
“Supersekundærstruktur” (“motifs”) • Kombinasjoner av påfølgende sekundær-strukturelementer (SSEer), f eks bab, b-hårnål, aa, b-tønne
Protein struktur og folding • For å bli til et funksjonelt protein må den • nysyntetiserte aminosyrekjeden få en tertiær • struktur. Denne strukturen må være: • Tilstrekkelig stabil • Oppnåelig Oppnåelig vil si: • Det må finnes en folding ”pathway” som fører til at strukturen blir dannet • ”Off-pathway” prosesser (f eks utfelling / aggregering) må motarbeides
Chaperones (ikke pensum) Chaperoner og chaperoniner beskytter mot ”off-pathway” prosesser, f eks GroEL/ES systemet [bildet er fra Science 284:822-825 (1999)]
Protein folding (side 224-225) The Levinthal paradox is more or less solved: Local (secondary) interactions and an intrinsic tendency of unfolded proteins to form (transient) elements of secondary structure steer the protein into a ”folding funnel”. D. Baker, Nature 405:39-42 (2000)
Protein folding (side 224-225) • Three steps: Transient elements of secondary structure Hydrophobic collapse, sekundær struktur dannelse Completion of folding (tertiary interactions) The rate-limiting step is between phase 2 and 3 and is the same for all molecules Names for this mechanism: ”nucleation condensation” (Fersht), ”extended nucleus”, framework model (Kim & Baldwin)
Protein struktur og stabilitet (side 221- 224) • For (nesten) hvert protein finnes det en unik struktur som er den energetisk mest gunstige. • Stabilitet er marginal og er summen av store negative effekter og store positive effekter av folding • Negativ effekt av folding: tap av entropi • Positiv effekt av folding: hydrofob effekt • Det finnes mange ”restraints” som begrenser hva som er mulig i naturen (se side 223-224; eksempel: Ramachandran plott)
PROTEIN STRUKTUR: • Eksperimentelle metoder • Databaser • Visualisering
Eksperimentell bestemmelse av proteinstruktur • Lav-resolusjons teknikker: • Circulær dikroisme sepktroskopi (sekundær struktur, stort sett ”all-or-nothing”) • Fluorescence (tertiær struktur – ”all-or-nothing”) • Elektronmikroskopi (brukes av og til for å få et lav-resolusjonsbilde av store proteinkomplekser) • Röntgenkrystallografi • Proteinet må krystalliseres • Elektrontettheten bestemmes • Atomkoordinatene utledes • Gir statiske modeller • Bottle neck: krystallisering • NMR • Analyserer kjernespinnsresonnans • Utleder (flere) modeller som er konsistente med resonnansmønsteret • Viser reell variasjon for peptider i løsning, men også alternative modeller man ikke kan skille mellom • Bottle neck: complexity, solubility, labour power (but things are improving!)
Experimentell struktur bestemmelse • Gir oss strukturdatabasen som er grunnlaget for mange prediksjonsmetoder • Tar mye mer tid enn ”bioinformatisk struktur bestemmelse” (f. eks. modell bygging) • Men ”bioinformatisk struktur bestemmelse” er ikke alltid mulig og gir dessuten mindre nøyaktige resultater
Hvordan beskriver man en struktur? • Kjemisk sammensetning • Forbindelser mellom atomene (”chemical connectivity”) • Atom koordinater, x, y og z • Vann molekyler og ligander • Chemical bonds in structures: • Chemistry rules approach: man bruker kjemiske regler for å rekonstruere bindinger i et bilde • Explicit bonding approach: all informasjon om ”bonds” ligger i koordinatfilen • Nb. ”Completeness”: strukturfiler er som oftest ikke komplett: • Små deler av proteinet mangler; i nesten alle krystalstrukturer • har man ikke protoner med
Mer avansert søk
Stadig flere strukturer -> en stadig bedre database for å oppnå forståelse og for å utvikle prediksjonsmetoder OBS! Mange ”redundant” structures……………
The Contents of a PDB File • HEADER: containing the file name and date. • TITLE: usually of a publication • COMPND: containing the name of the protein. • SOURCE: the organism from which the protein was obtained. • KEYWORDS • EXPDTA: method used • AUTHORS: persons who placed this data in the PDB • REVDAT: revision dates for data on this protein. • JRNL: relevant publications • REMARK: various types of information about the experiments, the file, symmetry, missing residues, quality checks (REMARK is usually many lines)
The Contents of a PDB File • DBREF: accession codes for this protein in other databases • SEQRES: explicit amino-acid sequence of the protein. • HET, HETNAM, FORMUL; information on cofactors, prosthetic groups, inhibitors or other nonprotein substances present in the structure. • HELIX, SHEET: elements of secondary structure in the protein. • LINK: contacts between heteroatoms and amino acids • CISPEP: peptide bonds in cis • SITE: information of binding sites and active sites • CRYST, ORIGX, SCALE: technical information on the coordinates, symmetry operations • ATOM and HETATM: atomic coordinate data
The Contents of a PDB File • SEQRES 8 129 VAL ASN CYS ALA LYS LYS ILE VAL SER ASP GLY ASN GLY 1HEW 67 • SEQRES 9 129 MET ASN ALA TRP VAL ALA TRP ARG ASN ARG CYS LYS GLY 1HEW 68 • SEQRES 10 129 THR ASP VAL GLN ALA TRP ILE ARG GLY CYS ARG LEU 1HEW 69 • HET NAG 201 15 N-ACETYL-D-GLUCOSAMINE 1HEW 70 • HET NAG 202 14 N-ACETYL-D-GLUCOSAMINE 1HEW 71 • HET NAG 203 14 N-ACETYL-D-GLUCOSAMINE 1HEW 72 • FORMUL 2 NAG 3(C8 H15 N1 O6) 1HEW 73 • FORMUL 3 HOH *103(H2 O1) 1HEW 74 • HELIX 1 A ARG 5 HIS 15 1 1HEW 75 • HELIX 2 B LEU 25 GLU 35 1 1HEW 76 • HELIX 3 C CYS 80 LEU 84 5 1HEW 77 • HELIX 4 D THR 89 ILE 98 1 1HEW 78 • HELIX 5 E VAL 109 ASN 113 1 1HEW 79 • SHEET 1 S1 2 LYS 1 PHE 3 0 1HEW 80 • SHEET 2 S1 2 PHE 38 THR 40 -1 N THR 40 O LYS 1 1HEW 81 OBS! Assignment av sekundær struktur er ikke triviell.
The Contents of a PDB File • The number and type of items/subjects in the header may vary. • The PBD keeps the actual X-ray data, the BioMagRes bank (Wisconsin) keeps the data for NMR structures • Keep in mind that the entry names (4 characters) are not ”logical” (that is usually not like e.g. ”8tln” for thermolysin) • See also in the book, page 125 – 131.