Download

1 / 37
410 likes | 903 Views
DEFECTOS DEL CICLO DE LA UREA. Dr. Javier Gonzálvez Aracil MIR R-1 Análisis Clínicos. ÍNDICE. Introducción: ciclo de la urea. Fisiopatología y clínica. Hiperamoniemia y deficiencias enzimáticas y de transportadores.

E N D
DEFECTOS DEL CICLO DE LA UREA Dr. Javier Gonzálvez Aracil MIR R-1 Análisis Clínicos
ÍNDICE • Introducción: ciclo de la urea. • Fisiopatología y clínica. • Hiperamoniemia y deficiencias enzimáticas y de transportadores. • Diagnóstico y seguimiento: métodos, determinación de amonio, aminoácidos y diagnóstico genético. • Tratamiento. • Conclusiones.
Introducción • Definición: Los errores congénitos del metabolismo (ECM) son entidades de una baja prevalencia individual, pero que colectivamente afectan a un número importante de individuos, ya que hasta la fecha se han descrito más de 1.000 diferentes. • Es importante que ante un cuadro clínico sugestivo, se establezca una hipótesis diagnóstica para iniciar estudios bioquímicos adecuados. • Los defectos del ciclo de la urea (DCU) son un grupo de ECM que debutan generalmente con hiperamoniemia y alteración del perfil de aminoácidos plasmáticos y urinarios. • La determinación de amonio es una prueba presente en la mayoría de laboratorios y es importante conocer y estas entidades a la hora de interpretar sus resultados y orientar futuros estudios.
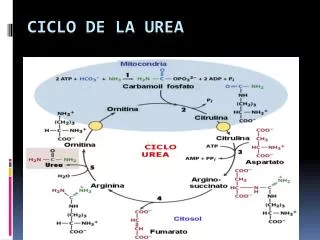
Introducción Ciclo de la urea: • Mediante el ciclo de la urea (CU) los mamíferos transforman el amonio, tóxico pata el SNC, en urea, que se elimina por la orina. • El amonio procede de la desaminación de las proteínas, de la dieta o edógenas, en caso de situación catabólicas. • Existen 9 defectos descritos relacionados con este ciclo, que afectan a seis enzimas y tres transportadores. La hiperamoniemia es su característica principal, y da lugar a los signos neurológicos por intoxicación del SNC. • El CU completo ocurre en el hígado. Hepatopatías pueden dar lugar a la misma clínica.
Introducción Ciclo de la Urea: • El CU fue descito en 1932 por Krebs y Henseleit. Consiste en 6 reacciones localizadas entre el citosol y la mitocondria. El balance global de cada vuelta es: • 2 NH+4 + HCO-3 + 3 ATP + H2O -> Urea + 2 ADP + AMP
Los seis enzimas implicados en la vía son: - la N-acetilglutamatosintetasa (NAGS, EC 2.3.1.1), - la carbamilfosfato sintetasa-1 (CPS-1, EC 6.3.4.16), - la ornitinatranscarbamilasa (OTC, EC 2.1.3.3), - la argininosuccinato sintetasa-1 (ASS-1, EC 6.3.4.5), - la argininosuccinato liasa (ASL EC 4.3.2.1) - y la arginasa (EC 3.5.3.1) Introducción
DCU: Fisiopatología y clínica • LOS DCU son uno de los grupos de ECM más comunes y con una incidencia global de 1 caso por cada 8.200 recién nacidos, siendo la deficiencia de OTC la que presenta una mayor prevalencia, y los déficits de NAGS y arginasa los de menor • La edad de debut es muy variable pero son tratables. • El defecto de cualquiera de estos enzimas provoca una acumulación del aminoácido(aa) sustrato, y una disminución del aa producto. • El exceso de amonio provoca un aumento secundario de glutamina, glicina y alanina.
DCU: Fisiopatología y clínica Fisiopatología(1): • La sintomatología viene condicionada por la hiperamoniemia y la hiperglutaminemia. • El mecanismo exacto es impreciso, siendo varios factores los que determinan su neurotoxicidad. • El acúmulo de amonio en el cerebro causa un incremento de la permeabilidad de la barrera hematoencefálica, una depleción de los intermediarios energéticos y una disgregación de los microtúbulos de las neuronas .
DCU: Fisiopatología y clínica Fisiopatología(2): • Con altas concentraciones mantenidas de amonio, se observan alteraciones en el desarrollo axonal y alteraciones en el metabolismo de neurotransmisores cerebrales, principalmente glutamato. • La importancia de la glutamina viene por ser precursor del amonio, de manera que aumentos de ésta preceden la hiperamoniemia. • Otras alteraciones de los DCU son de afectación hepática, dónde se observa fibrosis periportal en el déficit de OTC y la ASS-1junto a una elevación de las transaminasas y disminución del índice de protrombina.
DCU: Fisiopatología y clínica ´Clínica: forma neonatal: • Es la forma clásica ,corresponde al 60% de los casos, de comienzo agudo y debido a déficits muy graves. • Se inicia a las 24-72 horas de vida tras la ingesta de leche, con un cuadro clínico de encefalopatía aguda y progresiva, de rápida evolución. • Puede llegar al coma dependiendo de las concentraciones de amonio. • Existe una asociación entre la duración de la hiperamoniemia neonatal, el daño cerebral y la función intelectual futura.
DCU: Fisiopatología y clínica Clínica: forma de presentación tardía • Cuando existe una actividad residual de los enzimas. • En lactantes y niños es un cuadro poco específico tras una ingesta muy proteica o un aumento del catabolismo. Se caracteriza por problemas alimentarios, vómitos y encefalopatía, con hipertransaminasemia e hiperamoniemia. • En adolescentes y adultos la sintomatología es neurológica (migraña, disartria, ataxia) o psiquiátrica (alucinaciones, trastornos del comportamiento, discapacidad intelectual y trastornos del espectro autista).
Hiperamoniemia y deficiencias enzimáticas y de transportadores.
Hiperamoniemia y deficiencias enzimáticas y de transportadores Hiperamoniemia • Elevación de las concentraciones de amonio (> 150 µmol/L neonatos y > 80 µmol/L en edades posteriores) • Puede deberse a causas primarias o secundaria a otros trastornos • Hiperamoniemias primarias • Defectos de los enzimas del ciclo de la urea: NAGS, CPS-1, OTC, ASS-1, ASL y arginasa (puede cursar con valores normales de amonio). • Defectos del transporte de metabolitos relacionados con el ciclo de la urea: síndrome de hiperornitinemia-homocitrulinuria-hiperamoniemia (síndrome HHH), déficit de citrina y lisinuria con intolerancia a proteínas (LIP).
Hiperamoniemia y deficiencias enzimáticas y de transportadores • Hiperamoniemias secundarias(1) • Causas genéticas • Acidurias orgánicas: se acumulan ácidos orgánicos que causan una inhibición del activador inicial del ciclo, NAGS. • Deficiencia de la piruvato carboxilasa, reduce del aporte de aspartato. • Deficiencia de la piruvato deshidrogenasa, deficiencias de la β-oxidación de los ácidos grasos y deficiencia de carnitina, que originan baja disponibilidad de acetil CoA.
Hiperamoniemia y deficiencias enzimáticas y de transportadores • Hiperamoniemias secundarias(2) • Causas adquiridas • Hiperamoniemia transitoria del prematuro o recién nacido debido a inmadurez hepática. • Síndrome de Reye. • Tratamiento con valproato. • Malnutrición. • Derivaciones e insuficiencias hepáticas. • Quimioterapia para canceres hematológicos. • Infecciones del aparato urinario por bacterias ureolíticas.
Hiperamoniemia y deficiencias enzimáticas y de transportadores
Hiperamoniemia y deficiencias enzimáticas y de transportadores • Deficiencias enzimáticas(1): • Deficiencia de NAGS: cuadro clínico y bioquímico similar al de CPS-1. • Deficiencia de CPS-1: cataliza una reacción irreversible, de alto coste energético. Muy infrecuente y severo, incluso letal. • Deficiencia de OTC: ligada al X. Muy grave en varones. Las mujeres pueden desarrollar en algún momento hiperamoniemia e instintivamente adoptan una dieta baja en proteínas.
Hiperamoniemia y deficiencias enzimáticas y de transportadores • Deficiencias enzimáticas(2): • Deficiencia de ASS-1(citrulinemia tipo1): Hiperamonemia neonatal muy grave, fatal si no es detectada. Hay formas parciales. • Deficiencia de ASL(aciduria arginosuccínica): retraso en el desarrollo y sintomatología neurológica. El acúmulo del sustrato causa hepatopatía. • Deficiencia de arginasa(argininemia): infrecuente. No cursa con los síntomas habituales sino con diplejía espástica, retraso psicomotor y convulsiones. Las elevaciones de amonio son menores
Hiperamoniemia y deficiencias enzimáticas y de transportadores • Deficiencias de transportadores relacionados con el ciclo de la urea • Síndrome HHH (Hiperornitinemia-Homocitrulinuria-Hiperamoniemia): La clínica es más leve pero parecida al resto de defectos del CU. • Déficit de citrina (citrulinemia tipo II): afectación cerebral muy similar alresto de errores del CU. • Lisinuria con intolerancia a las proteínas (LIP): trastorno en el paso de los aa dibásicos (lisina, arginina y ornitina) a través de la membrana de las células epiteliales intestinales y renales. No interviene directamente en el CU, pero su alteración conlleva un desequilibrio de los sustratos. La clínica es parecida a los otros DCU.
Diagnóstico y seguimiento: métodos, determinación de amonio, aminoácidos y diagnóstico genético.
Diagnóstico • Algoritmo diagnóstico.
Diagnóstico • Pueden estar infradiagnosticadas al confundirse en neonatos con procesos más usuales como sepsis o hemorragia interventricular. • Se debe sospechar ante el cuadro habitual: neonato que, a las 24 o 48 horas presenta encefalopatía aguda, progresiva y de rápida evolución. • En mayores, alteraciones de índole neurológico. • Se deberá determinar el amonio y otras pruebas hematológicas y bioquímicas. • Confirmada la hiperamoniemia, seguir el algoritmo. • En la mayoría de los programas de cribado neonatal se incluye la citrulinemia tipo 1 y al aciduria arginosuccínica. • El seguimiento bioquímico de los afectos se realiza mediante controles diarios de amonio y aa hasta su normalización y después controles periódicos en función de la evolución y otros parámetros.
Diagnóstico • Métodos: • Para la determinación de amonio existen diversos métodos: colorimétrico, fluorimétrico, detección por electrodo o enzimático, siendo el enzimático con glutamato deshidrogenasa el más utilizado. • Esta enzima cataliza la conversión de amonio más α-cetoglutarato a glutamato con la oxidación de NADPH a NADP+.La medida de la disminución de absorbancia a 340 nm es proporcional a la concentración de amonio. • En el resto de las determinaciones empleadas se aplican técnicas que se realizan en laboratorios especializados.
Diagnóstico • Determinación de amonio: • Es el signo bioquímico guía. Se considera hiperamonemia un valor mayor de 150 μmol/L durante el período neonatal, y mayor de 80 μmol/L posteriormente. • Es importante el aspecto preanalítico. • El retraso en el procesamiento de la muestra o una muestra hemolizada dará un valor elevado. • Si no se puede procesar de inmediato, el plasma debe ser congelado en el momento. • También se eleva con el ejercicio, fármacos y el tabaco. • Se considera de elección como anticoagulante el EDTA y que la extracción se realice en ayunas.
Diagnóstico • Determinación de aa y ácido orótico(1): • La citrulina plasmática se encuentra elevada en los déficits citoplasmáticos y en los de transporte, menos en el síndrome HHH, donde está ligeramente disminuida. • El déficit de arginina plasmática es común a todos los defectos primarios excepto en el déficit de arginasa, en el que su valor se eleva. • Las concentraciones elevadas de glutamina.(800-1.000 μmol/L; valores de referencia < 730 μmol/L), suelen aparecer en todos los DCU al debut y en las descompensaciones. • La presencia en orina o plasma de ácido arginosuccínico y de sus derivados anhidros es el signo patognomónico de la aciduria arginosuccínica.
Diagnóstico • Determinación de aa y ácido orótico(2): • La elevación de la ornitina en plasma, junto a una hiperamoniemia y a la elevación de homocitrulina en orina, nos indicará un defecto del transportador ORNT-1 (síndrome HHH). • Un descenso plasmático de la lisina y su elevación urinaria, nos orientará hacia un diagnóstico de LIP. • El ácido orótico y la orotidina derivan del exceso de carbamilfosfato. Este acúmulo es básico para el diagnóstico de los trastornos que cursan con citrulina baja: OTC y CPS-1 (o NAGS). Si no presentara hiperamonemia, habrá que pensar en otros transtornos metabólicos.
Diagnóstico • La gran ventaja de la determinación de aminoácidos es que permite un diagnóstico precoz, permitiendo el inicio de determinados tratamientos específicos sin necesidad de esperar al diagnóstico genético • Aún así, el diagnóstico definitivo ES mediante estudio genético.
Tratamiento • Tratamiento de urgencia: • Está encaminado a disminuir la producción de amonio: supresión de la ingesta proteica (nunca más de 24-48 horas) y el aumento de aporte de glucosa y lípidos para evitar el catabolismo endógeno. • La utilización de fármacos quelantes (benzoato sódico, fenilbutirato sódico) favorecen la eliminación del amonio. • Si la concentración de amonio supera los 400 μmol/L, se recomienda detoxificación extracorpórea (hemofiltración, hemodiafiltración o diálisis peritoneal). • El tratamiento con L-arginina (facilita la activación del ciclo de la urea) y carnitina (favorece la eliminación de productos tóxicos y previene la deficiencia de esta sustancia) también es de uso general.
Tratamiento • Tratamiento de mantenimiento(1): • Está encaminado a evitar descompensaciones y la educación al paciente y su entorno un punto fundamental. El tratamiento debe ser individualizado, dependiendo de la tolerancia proteica del paciente. • Reducción importante de la ingesta de productos ricos en proteínas (carne, pescado, huevos, lácteos y legumbres). Esta dieta se complementa con alimentos ricos en proteínas de alto valor biológico o preparados de aminoácidos esenciales. • Los suplementos dietéticos utilizados son arginina, citrulina, arginina y vitaminas y elementos traza. • El aporte calórico debe mantenerse un 10-15 % por encima de las necesidades del paciente.
Tratamiento • Tratamiento de mantenimiento(2): • El uso de quelantes favorece la excreción de nitrógeno mediante distintos productos de la urea. • En casos de debut neonatal y difícil manejo, el trasplante hepático entre los 3 y los 12 meses de edad es la mejor opción.
Conclusiones • El ciclo de la urea está regido por seis enzimas y tres transportadores. • La deficiencia de alguno de estos nueve componentes resulta en un bloqueo del ciclo y en una elevación del amonio plasmático. • Estos defectos varían en severidad pero comparten clínica caracterizada por retraso en el desarrollo, retraso mental, convulsiones, vómitos, anorexia, letargia, estado mental alterado y coma. • El diagnóstico bioquímico se basa en la determinación en plasma de amonio y aminoácidos, y en orina de aminoácidos y ácido orótico. El estudio genético de la proteína defectuosa permite el diagnóstico definitivo. • El tratamiento consiste en controlar estrictamente los niveles del amonio, juntamente con un control dietético y farmacológico por parte de un equipo pluridisciplinar.