Download
1 / 18
220 likes | 805 Views
بیماری ویلسون. عرفان زراعت کار ۹۱۱۱۱۰۲۶. بیماری ویلسون یا hepatolenticular degeneration (فساد کبدی عدسی وار) یک بی نظمی ژنتیکی (مایل به بازگذشت اتوزومال) است که مس در بافت ها انباشته می شود.

E N D
بیماری ویلسون عرفان زراعت کار ۹۱۱۱۱۰۲۶
بیماری ویلسون یا hepatolenticulardegeneration (فساد کبدی عدسی وار) یک بی نظمی ژنتیکی (مایل به بازگذشت اتوزومال) است که مس در بافت ها انباشته می شود. این بیماری به وسیله ی علایم عصبی یا روانپزشکی و بیماری کبد آشکار و با روش دارویی که جذب مس را کاهش می دهد یا زیادی مس بدن را حذف میکند, درمان می شود ولی گهگاه پیونذ کبد نیاز است. این شرایط وابسته به جهش در ژن پروتئین بیماری ویلسون (ATP7B)است. یک کپی از ژن غیر معمول در یک درصد افراد مشاهده می شود که هیچ علامتی ندارند. علایم معمولا بین سن ۶ تا ۲۰ سالگی پدیدار می شوند. بین ۱ تا ۴ نفر از هر ۱۰۰۰۰۰ نفر دارای بیماری ویلسون است. این به بیماری به خاطر ساموئل الکسااندر کاینیر ویسلون (۱۹۳۷ - ۱۸۷۸) که یک نورولوژیست انگلیسی است که نخستین بار به توصیف این بیماری پرداخت (۱۹۱۲) نام گرفت.
علائم قسمت عمده ی ذخیره مس در کبد و مغز است.افراد با مشکل کبدی گرایش به درمان دارویی فوری دارند به ویژه وقتی که کودک یا نوجوان اند نسبت به فرد ۲۰ ساله یا بزرگ تر با علایم عصبی و روانپزشکی. برخی افراد به واسطه ی ارتباط خیشاوندی با یک بیمار شناسایی می شوند که بسیاری از این ها زمانی که تست می شوند مشخص می شود که علائم را تبرجه کردند ولی تشخیصی برایشان داده نشده است.
بیماری کبد • بیماری کبد به صورت خستگی ظاهر می شود و گرایش خونریزی را افزایش می دهد یا پریشانی و فشار خون پورتال می شود.سپس وضعیتی که در آن فشار در ورید پورتال به صورت نشانه دار افزایش میابد به واسطه ی واریس مری ‚ عروق خونی در مری به بزرگی طحال و انباشتگی مایع در حفره ی شکمی است. • در یک تست علامت بیماری کبد حاده همانند اسپایدر ناوی (spider naevi)ممکن است مشاهده شود.هپاتیت فعال حاد موجب سیروز کبد می شود که اغلب در طول زمان علامت دار می شود. • در حال ی که بیشتر مردم سیروز دار با خطر تومور بد خیم هپاتو سللولار مواجه اند‚این خطر در ارتباط کم با بیماری ویلسون است.تنها حدود پنج درصد مردم هنگامی که نارسایی حاد کبدی دارند تشخیص داده می شوند. • علائم روانپزشکی در ارتباط با علائم نورولوژیکال و به ندرت بازنمود می شود.علائم کمتر آشکار شده و بعضی اوقات به دلایل دیگر نسبت داده می شود.به این خاطر تشخیص بیماری ویلسون به ندرت است آن زمانی است که با علائم روانپزشکی باشد.
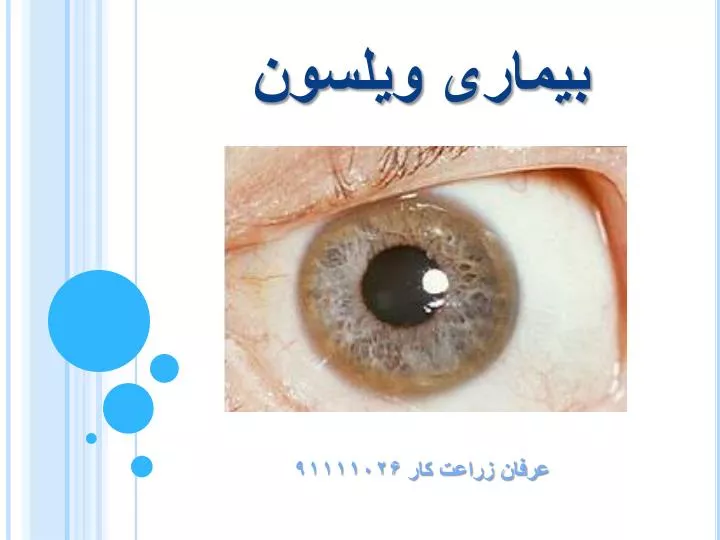
شرایط پزشکی در ارتباط با ذخیره ی مس در بیماری ویلسون • چشم:حلقه ی کایزر فلیشر(kayser-fleischer) که یک علامت شاخص است ممکن است در کورنه آد چشم مشاهده شود.این ها به خاطر رسوب مس در غشای دسمت (descemet) است که در همه بیماران ویلسونی مشاهده نمی شود.بیماری ویلسون همچنین در ارتباط با کاتاراکت سان فلاور (sunflower) که لا پیگمان های قهوه ای یا سبز در جلو و عقب کپسول لنز مشخص می شود و باعت کاهش دید مشخص است. حلقه ی کایزر-فیشر تقریبا در ۶۶٪ تشخیص هاست. • کلیه:اسیدوز رنال تیوبولار (renal tubular) یک اختلال با دخالت بیکربنات در تیوبول پروکزیمال در ارتباط نفروکلسینوزیس (nephrocalcinosis) و گهگاه آمینواسیدوریا (aminoaciduria) است. • قلب:کاردیومایوپبتی (cardiomyopathy) کمیاب است ولی تشخیص آن در ویلسون مشکل و ممکن است منجر به نارسایی قلبی و آریتمی قلبی شود • هورمون:هایپوپاراتیروئیدیسم (hypoparathyroidism) و نازایی (infertility) و سقط جنین (habitual abortion).pi
ژنتیک • ژن بیماری ویلسون (ATP7B) در کروموزوم شماره ١٣و به سرعت در کبد و کلیه و جفت آشکار است.کد ژن برایP-type ATPase مس را در صفرا و ترکیب در سرولوپلاسمین منتقل می کند.جهش می توتند در ۹۰٪ موارد شناسایی شود. ۶۰٪ هموزیگوس برای این ژن جهش یافته و ۳۰٪ دارای یک کپی غیر طبیعی و ١۰٪ بدون جهش اند.اگر چه ۳۰۰ جهش ژن توضیح داده شده‚در بیشتر جمعیت ها عامل ویلسون در ارتباط با تعداد کمی از جهش ها ویژه برای آن جمعیت است. • تغییر نرمال ژن PRNP میتواند عامل بیماری به واسطه ی تاخیر در سن وقوع و تاثیر نوع علامت ها و پیشرفت آن را اصلاح کند.این ژن پروتئین پرایون (prion) را ساخته که در مغز و بقیه ی بافت ها فعال و در انتقال مس دخالت دارد. • بیماری ویلسون بیشتر در گروه بیماری های ارثی است مه اضافه بار مس در کبد است که همگی موجب سیروز در جوانی می شود
پاتوفیزیولوژی • مس برای بعضی از عملکرد ها برای بدن لازم است,غالبا به عنوان کوفاکتور برای بعضی آنزیم ها مثل سرولوپلاسمین و سیتو کروم سی اکسیداز (cytochrome c oxidase) و ... . پروتئین منتقل کننده ی مس در روده ی کوچک (1CMT) حامل مس درون سلول است .برای عامل افزایش غلظت مس آنزیمی که ATP7A نامیده می شود مس را در ورید پورت آزاد می کند.سلول های کبد حامل پروتئین CMT1 و متالوتیونئین و ATOX1 متصل به آن در سلول است ولی در اینجا ATP7B موجب رهاسازی آن در گردش خون می شود که هر دو عمل آن در ویلسون معیوب است.ذخیره مس در بافت کبد:سرولوپلاسمین هنوز ترشح می شود ولی در این فرم کمبود مس و کم شدن سریع آن در گردش خون است.هنگامی مقدار مس کبد پایمال شود پرئتئینی که معمولا متصل به آن است موجب آسیب اکسیداتیو از طریق پروسه ای که فنتون کمیستری (fenton chemistry) نامیده می شود می گردد.
این آسیب سرانجام در ارتباط با هپاتیت حاد فعال و فیبروسیس و سیروز است.کبد همچنین مس را در گردش خون ترشح می کند تا به سرولوظلاسمین متصل نماند.این مس آزاد ناگهان سراسر بئن را گرفته مخصوصا در کلیه چشم و مغز.مغز بیشتر مس را در گانگلیای بازال (basal ganglia) سپرده مخصوصا در پوتامن (putamen) و گلوبوس پالیدوس (globuspallidus) این نواحی معمولا در هماهنگی انتقال شریک اند.آسیب این نواحی موجب علامت روانپزشکی در ویلسون می شود. • معمولا واضح نیست چرا ویلسون موجب همولیز می شود ولی مس آزاد تاثیر مستقیم در اکسید شدن هموگلوبین‚جلو گیری از آنزیم های انرژی زا در RBC یا آُیب مستقیم غشای سلولی.
تشخیص • بیماران ویلسونی اکثرا تست عملکرد غیر طبیعی کبد کمی مثل افزایش آسپارتات ترنس آمیناز و آلانین ترنس آمیناز و بیلیروبین دارند. اگر آسیب کبد مهم باشد آنگاه آلبومین ممکن است کاهش یابد به خاطر ناتوانی سلول های آسیب دیده ی کبد در تولید پروتئین.پروترومبین تایم (prothrombin time) ممکن است به خاطر ناتوانی کبد در تولید پروتئین لخته کننده خون افزایش یابد . سطح آلکالین فسفاتاز مرتبطا در ویلسون به خاطر نارسایی حاد کبد کاهش یافته.اگر علامت های نورولوژی موجود باشد MRI مغز آنرا نشان داده و هایپراینسیتیز (hyperintensities) در قسمتی از مغز که در زمینه T2 بازال گانگلیا نامیده می شود.تست کامل مرتبط بیمارس ویلسون وجود ندارد ولی سطح سرولوپلاسمین و مس خون به خوبی مقدار مس دفع کرده در ادرار در طول ۲۴ ساعته برای فهم مقدار بدن استفاده می شود.مقدار استاندارد در تست های ایده آل بیوپسی کبد (liver biopsy) نامیده می شود
سرولوپلاسمین • سطح سرولوپلاسمین در ۸۰ تا ۹۵٪ موارد کم است (<0.2 g/l). به سبب فاز پروتئین حاد در التهاب این سطح در افراد نرمال است. سرولوپلاسمین کم با بیماری منکیز (menkes disease) و آسرولوپلاسمینمسا (aceruloplasminemia) در ارتباط است ولی با ویلسون به ندرت در ارتباط است. ترکیب علایم نورولوژیکال و حلقه ی کایزر-فلیشر . سطح کم سرولوپلاسمین به شایستگی برای تشخیص ویلسون مطرح می شود.اگر چه در بسیاری از موارد تست های بیشتری نیاز است.
مس سرم و ادرار • مس سرم قاعدتا کم است ملی مس ادرار در بیماری ویلسون ارزیابی می شود.ادرار در ۲۴ ساعت جمع شده سپس با کوپر-فری (copper-free) متر مقدار بالای g/24h100 نشان دهنده ی بیماری ویلسون است. و مقدار بالایg/24h 40 قویا هشداد دهنده است.مس بالای ادرار منحصر به ویلسون نیست و در هپاتیت خود ایمنی (autoimmune hepatitis) و کولستاسیس (cholestasis) نیز اتفاق می افتد • تست پنیسیلامین (penicillamine) در کودکان مورد استفاده است که mg 500 دوز پنیسیلامین از راه دهان داده شده و ادرار در ۲۴ساعت جمع و اگر مقدار بیشتر از 1600 μg باشد مطمعنا نشان دهنده ی ویلسون است.این تست برای بزرگسالا در دسترس نیست.
نمونه برداری کبد • یک تحقیق نشان دهنده ی ویلسون است که تست ایده آل برداشتن مقدار کمی از بافت کبد از طریق نمونه برداری کبد است.این ارزیابی میکروسکپیکی برای درجه ای از استیتوسیس (steatosis) و سیروز می باشد و شیمی بافتی و معرف مس برای اندازه گیری سختی ذخیره ی مس استفاده می شود.مقدار 250 μg از مس بر گرم در بافت کبد خشک تائید کننده ی ویلسون است.گهگاه مقدار کمتری از مس پیدا شده که در آن مورد ترکیب یافته های بیوپسی و دیگر تست ها نشان دهنده ی تشخیص رسمی ویلسون است. • در مراحل اولیه ی بیماری بیوپسی استیتوسیس را نشان می دهد.در بیماری های پیشرفته تغییر مشاهده شده بسیار شبیه آنچه که در هپاتیت خودایمنی رخ میدهد مشاهده می شود.در بیماری های پیشرفته سیروز مهمترین یافته است. • روش های شیمی بافتی برای شناسایی مس متناقض و غیر قابل اعتماد است وانجام تنهای آن کم توجها برای یک تشخیص است.
درمان • رژیم غذایی:در واقع رژیم غذایی با مس کم قابل پیشنهاد است همراه با پرهیز از قارچ و آجیل شکلات و میوه های خشک شده و جگر و صدف خوراکی • دارو:دارودرمانی برای ویلسون در دسترس است.بعضی مس بدن را کاهش می دهند در حالی که بقیه از جدب مس جلوگیری می کنند.عموما پنیسیلآمین (penicillamine) نخستین دارو است که به مس متصل و باعث دفع آن در ادرار است.از این رو مشاهده ی مقدار مس ادرار برای اندازه گیری شایستگی مقدار دوز بالاتر است. پنیسیلامین بدون عوارض نیست. حدود ۲۰٪ تجربه ی عوارض جانبی مثل سل جلدی (درد مفاصل و جوش پوستی) یا میاستنیا (myasthenia) و همراه آن علائم نورولوژیکی تقریبا نیمی از آنها با حاد شدن علائم اند.در حالی که این عارضه ها مشاهده می شود از ادامه پنیسیلامین جلوگیری و تراینتین هایدروکلراید (trientine hydrochloride) تجویز می شود.بعضی این دارو را ب عنوان نخستین راه درمان پیشنهاد ولی تاثیر پنیسیلامین وسیع تر است. تتراتایومولایبدیت (tetrathiomolybdate) اثرات سودمندی را در بعضی مطالعات نشان داده است. روی ممکن است در عوض از پایداری سطح مس در بدن حمایت کند.روی محرک متالوتایونئین (metallothionein) که یک پروتئین در سلول های روده که به مس جفت شده و از جذب و انتقال آن به کبد جلوگیری می کند.روی درمانی ادامه میابد مگر اینکه عود کند یا دفع ادراری مس افزایش یابد.
بسیار نادر است که راه های درمانی دهانی مخصوصا در بیماری های نورولوژیکال شدید جواب ندهند. دایمرکاپرول (dimercaprol) گهگاهی لازم است.این درمان به صورت تزریق عضلانی (intramuscularly) در چند هفته است و اثرات ناخوشایند مثل درد دارد. • افرادی که بدون علامتند (asymptomatic) عموما درمان می شوند به خاطر مس ذخیره شده ممکن است باعث آسیب دراز مدت در آینده شود.این موضوع هنوز مشخص نیست که این افراد بهتر است با پنیسیلامین یا روی استات درمان شوند.
درمان جسمی • تن درمانی برای بیماران با فرم نورولوژیک بیماری سودمند است.درمان مس ممکن است ۶ ماه براش شروع زمان ببرد و درمان جسمی می تواند همراه اتکشیا (ataxia) و دیستونیا (dystonia) و جنبش (tremors) به خوبی جلوگیری از پیشرفت انقباض (contracture) که نتیجه فرم دیستونیا است شود.
پیوند • پیوند کبد یک درمان تاثیرگذار برای ویلسون است ولی فقط در موارد خاص انجام می شود که به خاطر ریسک و پیچیدگی پروسه ی آن است که معمولا در افراد با نارسایی کامل کبدی که به درمان دارویی جوابی نمی دهند یا به شکل پیچیده ی بیماری حاد کبدی دچارند انجام می شود. از پیوند کبد در شکل سخت بیماری نیوروسایچایاتریک (neuropsychiatric) پرهیز می شود بدین خاطر که سود آن تظاهر نشده است.
رفرنس • Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML (2007). "Wilson's disease". Lancet 369 (9559): 397–408 • Kinnier Wilson SA (1912). Brain 34 (1): 295–507 • Merle U, Schaefer M, Ferenci P, Stremmel W (2007). • Lorincz MT (2010).Annals of the New York Academy of Sciences 1184: 173–87. • Yanoff, Myron; Jay S. Duker (2008). Ophthalmology (3rd ed. ed.). Edinburgh: Mosby. p. 411. • de Bie P, Muller P, Wijmenga C, Klomp LW (November 2007). J. Med. Genet. 44 (11): 673–88. • Grubenbecher S, Stüve O, Hefter H, Korth C (2006). "Prion protein gene codon 129 modulates clinical course of neurological Wilson disease". Neuroreport 17 (5): 549–52. • Roberts EA, Schilsky ML (2003) Hepatology 37 (6): 1475–92. • Lee, GR (1999). "Chapter 48: acquired hemolyticanaemias resulting from direct effects of infectious, chemical or physical agents". In Lee GR, Foerster J, Lukens J et al.. Wintrobe's clinical hematology. vol 1 (10th ed.). Williams & Wilkins. p. 1298. • Shaver WA, Bhatt H, Combes B (1986). "Low serum alkaline phosphatase activity in Wilson's disease". Hepatology 6 (5): 859–63 • Das SK, Ray K (September 2006).Nat ClinPractNeurol 2 (9): 482–93 • Walshe JM (July 1996). "Treatment of Wilson's disease: the historical background". QJM 89 (7): 553–5 • Brewer GJ, Askari FK (2005).Journal of Hepatology 42 (Suppl 1): 13–21. • Robertson WM (February 2000).Arch. Neurol. 57 (2): 276–7. • Cumings JN (1948). Brain 71 (Dec): 410–5. • McIntyre N, Clink HM, Levi AJ, Cumings JN, Sherlock S (February 1967). "Hemolyticanemia in Wilson's disease". N. Engl. J. Med. 276 (8): 439–44. • Cumings JN (March 1951). "The effects of B.A.L. in hepatolenticular degeneration". Brain 74 (1): 10–22. • Denny-Brown D, Porter H (December 1951). "The effect of BAL (2,3-dimercaptopropanol) on hepatolenticular degeneration (Wilson's disease)". N. Engl. J. Med. 245 (24): 917–25. • Vilensky JA, Robertson WM, Gilman S (September 2002). "Denny-Brown, Wilson's disease, and BAL (British antilewisite [2,3-dimercaptopropanol])". Neurology 59 (6): 914–6. • Walshe JM (January 1956). "Wilson's disease; new oral therapy". Lancet 267 (6906): 25–6.
Walshe JM (March 1982). "Treatment of Wilson's disease with trientine (triethylenetetramine) dihydrochloride". Lancet 1 (8273): 643–7. • Harper PL, Walshe JM (December 1986). "Reversible pancytopenia secondary to treatment with tetrathiomolybdate". Br. J. Haematol. 64 (4): 851–3. • Brewer GJ (January 2000)Proc. Soc. Exp. Biol. Med. 223 (1): 39–46. • Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993). "The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene". Nat. Genet. 5 (4): 327–37. • Tanzi RE, Petrukhin K, Chernov I, et al. (1993). "The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene". Nat. Genet. 5 (4): 344–50. • Sternlieb I, Twedt DC, Johnson GF, et al. (1977). "Inherited copper toxicity of the liver in Bedlington terriers". Proc. R. Soc. Med. 70 Suppl 3 (Suppl 3): 8–9. • van De Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002). • Müller T, van de Sluis B, Zhernakova A, et al. (2003). "The canine copper toxicosis gene MURR1 does not cause non-Wilsonian hepatic copper toxicosis". J. Hepatol. 38 (2): 164–8.