Download

1 / 36
440 likes | 1.48k Views
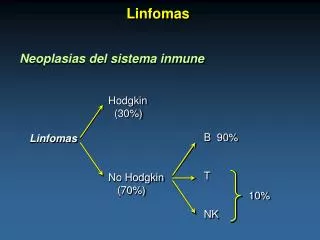
Linfomas. Introdução Linfoma de Hodgkin. Thomas Hodgkin (1798-1866). Sistema linfático. Introdução Linfoma de Hodgkin. Raro em crianças; Incidência maior em adultos jovens; Predomínio no sexo masculino. Introdução Linfoma de Hodgkin.

E N D
IntroduçãoLinfoma de Hodgkin Thomas Hodgkin (1798-1866)
IntroduçãoLinfoma de Hodgkin • Raro em crianças; • Incidência maior em adultos jovens; • Predomínio no sexo masculino.
IntroduçãoLinfoma de Hodgkin • As células que dão origem aos linfomas são originárias de células maduras. Colonizam os órgãos linfóides secundários podendo acometer outros órgãos, tais como pulmão, ossos, cérebro; • O linfoma de Hodgkin é caracterizado pela presença de células de Reed-Sternberg (RS) em cortes histotógicos de tecidos afetados pela doença.
Célula de Reed-Sternberg (RS)Linfoma de Hodgkin Multinucleada, com aspecto de olho de coruja. O citoplasma é bem abundante. Originárias de linhagem de linfócitos B
Linfoma de Hodgkin • As células encontradas no linfoma de Hodgkin, além das células RS, são células mononuclerares com marcação CD30+(receptor de proliferação de células T e B) e CD15+ (trissacarídeo terminal), sem expressão de antígenos da linhagem B; • Ocorre intensa resposta inflamatória, com a presença de muitas células normais reativas.
Linfoma de Hodgkin • Estadiamento clínico: através de exames de imagem (RX, TC, PET-TC) Pré-Qt Pós-Qt
EstadiamentoLinfoma de Hodgkin • Estádio I: envolvimento de linfonodos de 1 quadrante acima do diafragma; • Estádio II: envolvimento de mais regiões linfonodais acima do diafragma; • Estádio III: envolvimento de regiões linfonodais acima e abaixo do diafragma; • Estádio IV: além do envolvimento linfonodal, acometimento de outros órgãos (linfóides ou não).
EstadiamentoLinfoma de Hodgkin Todos os estádios são ainda classificados como A(ausência) ou B(presença), de acordo com a presença dos seguintes sinais ou sintomas: • Febre acima dos 38°C; • Sudorese noturna; • Perda de mais de 10% do peso em 6 meses.
Sinais e sintomasLinfoma de Hodgkin • Linfadenopatia assimétrica indolor de consistência firme e emborrachada; • Esplenomegalia (indicativo de disseminação hematogênica; • Comprometimento mediastinal com ou sem derrame pleural; • Fraqueza, anorexia, fadiga, prurido; • Anemia normocítica e normocrômica; • Leucopenia com perda de imunidade celular.
TratamentoLinfoma de Hodgkin • Radioterapia • Quimioterapia
IntroduçãoLinfoma não-Hodgkin • Engloba um grande número de neoplasias linfóides, na maioria de linhagem B, com evoluções clínicas bem variadas; • Apresentam disseminação irregular, inclusive extranodal.
ClassificaçãoLinfoma não-Hodgkin Órgão linfóide secundário Centro germinativo Zona do manto Zona marginal Células B originárias da medula óssea não-estimuladas. Ativação celular com mudança de classes de isótipos de Igs. Ocorrência de mutações.
ClassificaçãoLinfoma não-Hodgkin Linfoma folicular Mieloma Linfoma de células do manto Zona folicular (B) Zona do manto (T) Zona marginal Linfoma linfocítica crônica de células B Linfoma da zona marginal Linfoma linfoplasmocitóide Linfoma difuso de células grandes
ClassificaçãoLinfoma não-Hodgkin • Baixo grau: linfoma linfocítico, linfoplasmocitóide, linfoma da zona marginal, folicular e das células do manto. • Alto grau: linfoma difuso de células grandes, linfoma de Burkitt (associado à infecção por EBV); linfomas de células T: associado à infecção por HTLV, micose fungóide e síndrome de Sézary.
Sinais e sintomasLinfoma não-Hodgkin • Similares aos sintomas e sinais do Linfoma de Hodgkin, podendo-se apresentar mais disseminado para órgãos não-linfóides.
TratamentoLinfoma não-Hodgkin • Radioterapia • Quimioterapia
Mieloma múltiplo Profª Heide Baida
Introdução É uma neoplasia derivada de plasmócitos. Caracteriza-se pela presença de uma imunoglobulina monoclonal no soro dos pacientes. Esta é conhecida como PARAPROTEÍNA (PEOTEÍNA M)e na eletroforese de proteínas plasmáticas é responsável pela presença da banda M.
Introdução BANDA M ALBUMINA BETA ALFA2 GAMA ALFA1
Introdução • Responsável por cerca de 1% dos cânceres; • Atinge cerca de 10% de todos os cânceres hematológicos; • Comum entre indivíduos da raça negra; • Acomete mais os idosos acima dos 65 anos.
Introdução Etiopatologia: Proliferação progressiva e descontrolada de plasmócitos na medula óssea, podendo formar tumores (plasmocitomas) únicos ou múltiplos, mas geralmente apresentam a forma disseminada. As células neoplásicas invadem tecido ósseo, provocando lesões osteolíticas, principalmente nos ossos chatos. Pode ainda provocar osteoporose difusa, com dores ósseas e fraturas patológicas.
Estadiamento • I: cálcio normal, ausência de lesões ósseas disseminadas, Igs normais. • II: intermediária e variável. • III: cálcio aumentado, lesões ósseas líticas, Igs aumentadas, inclusive em urina de 24 hs.
Sinais e sintomas • O envolvimento ganglionar é raro; • Dores ósseas; • Insuficiência renal; • Fraqueza, letargia, dispnéia, palidez;
Imagens radiográficasLesões líticas conhecidas como “saca bocados”
Laboratorialmente • Hipercalcemia; • Anemia normocítica e normocrômica; • Eletroforese de proteínas com pico de gamaglobulinas (em 80% dos casos); • Proteína monoclonal, geralmente das classes IgG (50%) ou IgA (20%) • Proteinúria com presença da proteína de Bence-Jones: composta por cadeias leves oriundas do soro; • Medula óssea rica em plasmócitos.
Plasmócito Plasmócitos (medula óssea)
Tratamento: • Quimioterapia; • Radioterapia; • Talidomida; • TMO alogênico (em alguns casos); • TCT
Gamopatia monoclonal de origem indeterminada (GMOI) São patologias, neoplásicas ou não, que podem levar à diagnósticos errados. São as principais: • Doenças dos colágeno; • Hepatite B ou Hepatite C; • Tumores sólidos; • Doenças mieloproliferativas; • Linfoma de Hodgkin; • Casos de TMO alogênico; • Casos de transplantes de órgãos.
Obrigada! Universidade Nove de Julho