Download
1 / 41
420 likes | 791 Views
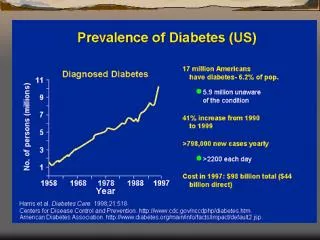
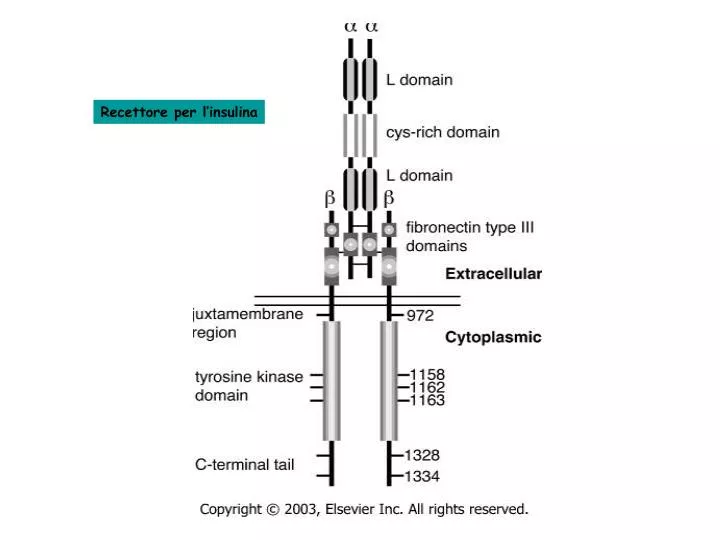
Recettore per l ’ insulina. Muscolo scheletrico. Fegato, muscolo sch. Attivazione glicogenosintesi. Inibizione gluconeogenesi. Attivazione lipogenesi. Tess. adiposo. fegato. Casi di diabete (in milioni) in diverse parti del mondo. DIFETTI DI. SECREZIONE DA PARTE. DI. CELLULE. b.

E N D
Muscolo scheletrico Fegato, muscolo sch. Attivazione glicogenosintesi Inibizione gluconeogenesi Attivazione lipogenesi Tess. adiposo fegato
DIFETTI DI SECREZIONE DA PARTE DI CELLULE b PANCREATICHE DIFETTI DI AZIONE DELL’INSULINA DIABETE DI TIPO I: distruzione autoimmune delle b cellule pancreatiche DIABETE DI TIPO II: gruppo eterogeneo di alterazioni caratterizzato da diversi gradi di resistenza all’insulina, diminuita secrezione di insulina,aumentata produzione di glucosio.
Diminuita inibizione della sintesi di glucosio in risposta all’insulina a livello del fegato Diminuito uptake di glucosio in risposta all’insulina a livello del muscolo scheletrico Diminuita inibizione della lipolisi in risposta all’insulina a livello del tessuto adiposo
Vi sono diverse isoforme di IRS che sono espresse differenzialmente in diversi tessuti Taniguchi et al.Nature Reviews Molecular Cell Biology7, 85–96 (February 2006) | doi:10.1038/nrm1837
Quale tipico nodo critico del segnale insulinico, IRS è bersaglio di segnali In grado di regolare negativamente la risposta all’insulina SEGNALE Protein Tyrosine Phosphatase 1B (PTP1B) (tirosin fosfatasi) fosforilazione in serina da parte di diverse Ser/Thr chinasi inibisce il segnale Taniguchi et al.Nature Reviews Molecular Cell Biology7, 85–96 (February 2006) | doi:10.1038/nrm1837
3 2 4 1 Taniguchi et al.Nature Reviews Molecular Cell Biology7, 85–96 (February 2006) | doi:10.1038/nrm1837
Inibita da fosforilazione Attivata da fosforilazione Glicogenolisi e glicogeno sintesi sono regolate da diversi ormoni e mediante segnali che esercitano azione opposta sugli enzimi limitanti queste due vie metaboliche Tratta da Marks et al. “Cellular Signal Processes”, Garland Science
2. glicogenosintesi Ramificazione/Deramificazione Glicogeno Glicogeno Fosforilasi b (defosforilata). Meno attiva Glicogeno sintasi I PP1 AMPc/PKA Glicogeno sintasi D (fosforilata) Meno attiva Glicogeno Fosforilasi a (fosforilata). Attiva Glucosio-1-P Fosfoglucomutasi Glucosio-6-P Glucosio-6-Pasi Glucosio
Insulina Akt/PKB - Glicogeno sintasi Chinasi (GSK3) Glicogeno Glicogeno sintasi I PP1 (Protein Phosphatase 1) Glicogeno sintasi D (fosforilata) Meno attiva Glucosio-1-P Glucosio-6-P Glucosio 2. glicogenosintesi
La fosforilazione della glicogeno sintasi (e la sua conseguente inibizione) non è mediata solo da GSK3, ma anche da altre chinasi che agiscono in modo coordinato Attivata da AMPc generato da Segnali b-adr e glucagone Fosforilata e inibita da insulina (mediante AKT/PKB) Costitutivamente attiva, consente fosforilazione da parte di PKA Costitutivamente attiva, consente fosforilazione da parte di GSK3 e AMPK Attivata da aumento AMP conseguente a riduzione ATP (sensore di livello energetico) Tratta da Marks et al. “Cellular Signal Processes”, Garland Science
Phosphoenolpyruvate caboxykinase “acetyl-CoA carboxylase” 3. Inibizione gluconeogenesi “fatty acid sinthase” In blue enzimi glicolitici e favorenti la lipogenesi. L’insulina aumenta la trascrizione dei geni codificanti per questi enzimi In rosso enzimi implicati nella gluconeogenesi. L’insulina inibisce la trascrizione dei geni codificanti per questi enzimi In verde enzimi la cui attività è regolata dall’insulina attraverso modificazioni del loro stato di fosforilazione
3 2 4 1 Taniguchi et al.Nature Reviews Molecular Cell Biology7, 85–96 (February 2006) | doi:10.1038/nrm1837
Attivano TSC2 e quindi inibiscono mTOR Inibiscono TSC2 e quindi attivano mTOR RAS GAPs Tuberina* GTP-binding protein Amartina* mTORC1 Fattore iniziante la traduzione * Mutate in una sindrome di tumori famigliari (“sclerosi tuberosa”): tumori benigni diffusi chiamati amartomi a livello di rene, polmone, cervello e cute.
L’ipoglicemia e la carenza di nutrienti e ossigeno regolano positivamente l’attività di un bersaglio della via PI3K/AKT (inibendo mTORC1) ATTIVAZIONE TSC2 E INIBIZIONE mTORC1 mTORC1
Regolazione e funzioni di AMPK metformina sintesi proteica glicogenosintesi Liposintesi e sintesi colesterolo Aumenta espressione Glut4 e trasporto glucosio (mediante fosforilazione AS160) Aumenta lipolisi e glicolisi Tratta da Marks et al. “Cellular Signal Processes”, Garland Science
Sono molti i fattori che, in gran parte indirettamente (attraverso LKB1), attivano AMPK, tra cui dei farmaci (biguanidi: metformina ed altri) usati per il trattamento del diabete di tipo 2
AMP chinasi regola positivamente autofagia/mitofagia e biogenesi mitocondri, attraverso l’inibizione di mTORC1
Segnali a valle del recettore per l’insulina regolano l’invecchiamento cellulare In arancione proteine che inibiscono longevità. In verde proteine che promuovono longevità
Quale tipico nodo critico del segnale insulinico, IRS è bersaglio di segnali In grado di regolare negativamente la risposta all’insulina SEGNALE Protein Tyrosine Phosphatase 1B (PTP1B) (tirosin fosfatasi) fosforilazione in serina da parte di diverse Ser/Thr chinasi inibisce il segnale Taniguchi et al.Nature Reviews Molecular Cell Biology7, 85–96 (February 2006) | doi:10.1038/nrm1837
Obesità e ridotta sensibilità all’insulina: il tessuto adiposo diventa ipertrofico, va incontro a sofferenza e ciò innesta un processo infiammatorio (“adiposite”) ed il conseguente rilascio di citochine (TNF-a; IL-1b) che agiscono in loco (sull’adipocita) e a distanza (nel muscolo scheletrico e nel fegato) attivando chinasi (JNK, IKK) che fosforilano IRS inibendo la trasduzione del segnale insulinico, ma anche determinando una ridotta secrezione di insulina da parte delle cellule beta pancreatiche CON QUALI MECCANISMI FINORA DIMOSTRATI OBESITA’, IPERGLICEMIA E INFIAMMAZIONE INDUCONO INSULINO RESISTENZA??
1. ATTRAVERSO L’ADIPOSITE VERA E PROPRIA DANNO ADIPOCITARIO E RILASCIO DI CHEMOCHINE E CITOCHINE RECLUTAMENTO DI MONOCITI RILASCIO DI CITOCHINE CHE INIBISCONO IL SEGNALE INSULINICO
1. ATTRAVERSO L’ADIPOSITE VERA E PROPRIA DANNO ADIPOCITARIO E RILASCIO DI CHEMOCHINE E CITOCHINE LINFOCITI TH1/CD8+ + - RECLUTAMENTO DI MONOCITI LINFOCITI TH2/Treg RECLUTAMENTO E INDUZIONE MACROFAGI CON FENOTIPO M1 RILASCIO DI CITOCHINE CHE INIBISCONO IL SEGNALE INSULINICO
L’obesità induce una rapida alterazione del reclutamento di diversi tipi di linfociti T (vedi Nat Med 15 (agosto 2009), pag. 846) Mentre nel tessuto adiposo magro prevalgono linfociti Th2 e Treg (secernono IL-10 e inibiscono l’adiposite), nel tessuto adiposo di topi obesi si accumulano TH1 e CD8 che richiamano macrofagi e inducono infiammazione
1. ATTRAVERSO L’ADIPOSITE VERA E PROPRIA DANNO ADIPOCITARIO E RILASCIO DI CHEMOCHINE E CITOCHINE - - RECLUTAMENTO DI MONOCITI EOSINOFILI LINFOCITI TH2/Treg RECLUTAMENTO E INDUZIONE MACROFAGI CON FENOTIPO M2 RILASCIO DI CITOCHINE CHE INIBISCONO IL SEGNALE INSULINICO
2. ATTRAVERSO L’AZIONE DI ACIDI GRASSI RILASCIATI DAL TESSUTO ADIPOSO IPERTROFICO
Obesità e ridotta sensibilità all’insulina: prodotti del metabolismo di acidi grassi alterano il segnale insulinico mediante attivazione di Ser/Thr chinasi che fosforilano IRS e ne riducono l’associazione con Il recettore e la fosforilazione in tirosina. Fatty acid metabolism and insulin action in skeletal muscle or liver. Obesity results in an increased flux of free fatty acids into the circulation and uptake by the myocyte or hepatocyte. Activated fatty acids (i.e., fatty acyl-CoAs) are “metabolized” primarily via one of two pathways, oxidation or storage. When fatty acid flux exceeds the ability of these pathways to dispose of fatty acyl-CoAs, intermediaries of fatty acid metabolism (e.g., DAG, PA, LPA, ceramide) accumulate. In turn, these fatty acid intermediates can activate a number of different serine kinases that can negatively regulate insulin action. Ceramide can also impair insulin action through interactions with PKB/Akt. An inability to completely oxidize fatty acids through β-oxidation, which leads to an accumulation of acylcarnitines, has also been hypothesized to cause insulin resistance, although the precise mechanisms leading to insulin resistance are, to date, unknown. AGPAT, acylglycerol-3-phosphate acyltransferase; PAP, PA phosphohydrolase. . Clin. Invest. Simon Schenk, et al. 118:2992, 2008
3. ATTRAVERSO L’AZIONE DEL GLUCOSIO E DI ACIDI GRASSI CHE ATTIVANO L’INFLAMMOSOMA NLRP3 IN DIVERSI TIPI DI CELLULE COMPRESE LE CELLULE BETA PANCREATICHE INDUCENDO: -RILASCIO DI CITOCHINE -INIBIZIONE DI AMPK E INIBIZIONE DI MITOFAGIA
AMPK E INFLAMMOSOMA STANNO EMERGENDO COME POTENZIALI BERSAGLI DI TERAPIE ANTI-DIABETICHE
4. IPERALIMENTAZIONE, GLUCOTOSSICITA’, ACIDI GRASSI INDUCONO UPR E UPR INDUCE INFIAMMAZIONE PERK e IRE attivano NfkB e AP1 attraverso diversi meccanismi Tratto da Zhang e Kaufman, Nature 454:455, 2008